Step 4: Peak Lists
- Submitting Other Solvents or References
- Skipping peak list submission for specific compounds
- Autofilling Solvent, Frequency, and Temperature Information
- Validating
- Submitting
Peak List Formatting
This page is for filling out the peak values for H and C of your compound. This platform does not accept assignment information so you do not need to submit coupling information while submitting your list.
Peak List Formatting Rules
The same rules for filling out lists apply to both the C and H values. When submitting your peak lists, they must match the following formatting stipulations…
1) Peaks must be separated by commas or semicolons
-
Individual peak value must be separated each other by either commas “,” or semicolons “;”
-
Ex.
10.18, 11.70, 12.54, 13.2610.18; 11.70; 12.54; 13.26
2) Multiplets should be represented as ranges connected by dashes
- Multiplets can be represented as ranges containing the largest and smallest peak values. These ranges can be expressed by entering a dash “-“ in between the min and max values.
- i.e. if you have a doublet you should enter both peak values. If you have a quartet you should enter the highest and lowest values.
- Ex.
10.18, 11.83-12.22, 12.54, 13.44-13.65
3) You may include brackets surrounding peaks or ranges
- A single peak or range contained in round brackets is a valid input. Adding brackets can make your values easier to read.
- Brackets do not effect how the system interprets entered values.
- Ex.
10.18, (11.83-12.22), (12.54), 13.44-13.65
Obtaining A Peak List
1) Obtaining your peak list from MestReNova
Open your spectra in MestReNova software.
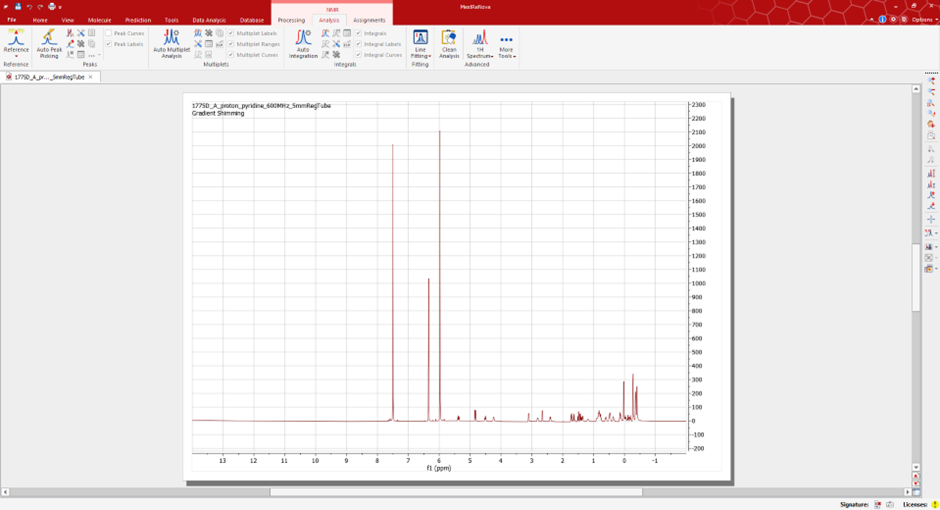
Under the NMR ‘Analysis’ tab, you will have the option to peak pick and interpret your data. Select the ‘Manual Peak Pick’ option and use the cursor to manually select the peaks of interest in your spectra.
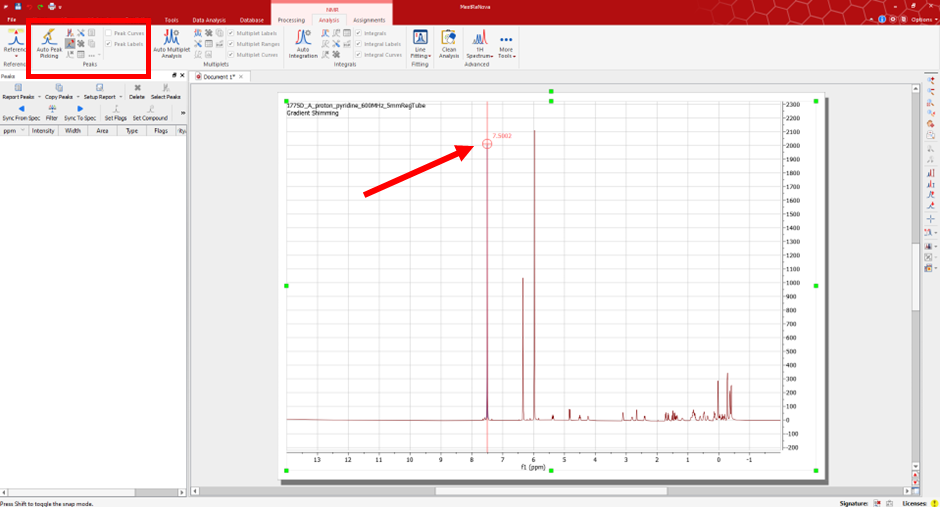
Once you have selected all the appropriate peaks you can generate the peak list. To do so, select the option ‘Report Peaks’.
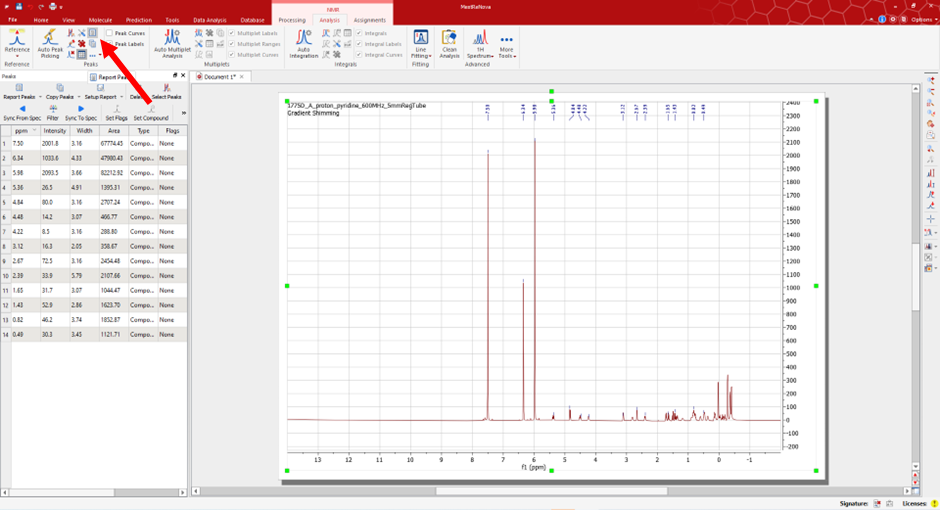
This will generate the list seen in the image, which is as follows:
1H NMR (600 MHz, pyridine) δ 7.50, 6.34, 5.98, 5.36, 4.84, 4.48, 4.22, 3.12, 2.67, 2.39, 1.65, 1.43, 0.82, 0.49.
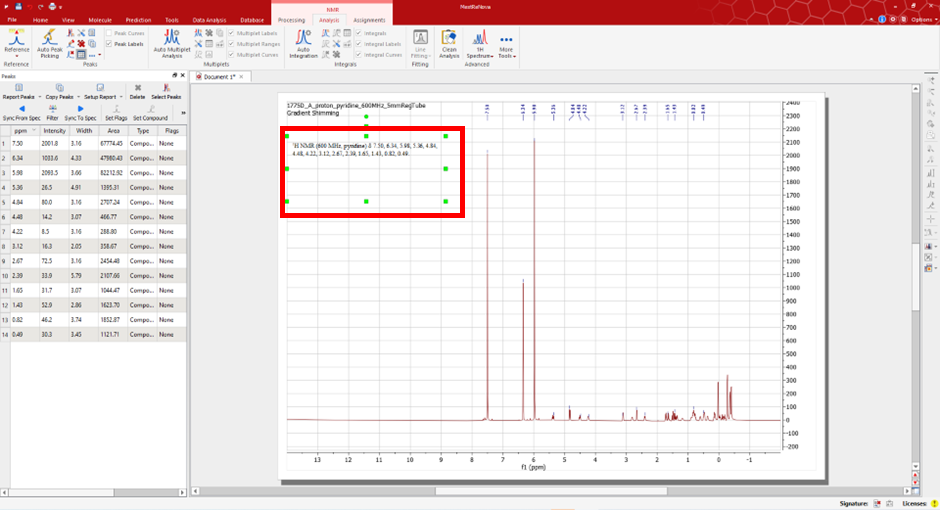
Remove the prefix metadata and submit only the peak values (comma separated) for your peak list. An example of an acceptable peak list can be seen below:
- Accepted peak list:
7.50, 6.34, 5.98, 5.36, 4.84, 4.48, 4.22, 3.12, 2.67, 2.39, 1.65, 1.43, 0.82, 0.49
Peak List Formatting Warnings and Errors
1) The number of values should not exceed the number of atoms
- i.e. If your structure has 4 unique Hydrogen atoms, the H peak list should not exceed 4 in length.
- Submitting excess values will throw an “Invalid number of atoms in peak list” error.
2) The values should be within a reasonable range for the type of atom they are referencing
-
Values outside of accepted ranges will throw an error and are not valid to submit.

-
i.e. values in your H peak list should not be less than -2 or exceed 20.
-
Values outside of expected ranges will throw a warning but are still valid to submit.

3) Lists must only contain valid characters
- The submitted peak list may only contain…
- Numerical digits
- The following characters: ( ) . , ; -
- Entering any other characters will throw an error.
4) A peak list may be submitted for only C or H
- You are not required to submit C and H peak lists for each compound.
- This will throw a warning, however, submitting a peak list for only one C or only H is valid.
If you have followed all the guidelines above then your peak lists should present green tables when you click “Validate”. Once all the peak lists present green, you can go ahead an click the “Submit” button.
Using The Peak List Interface
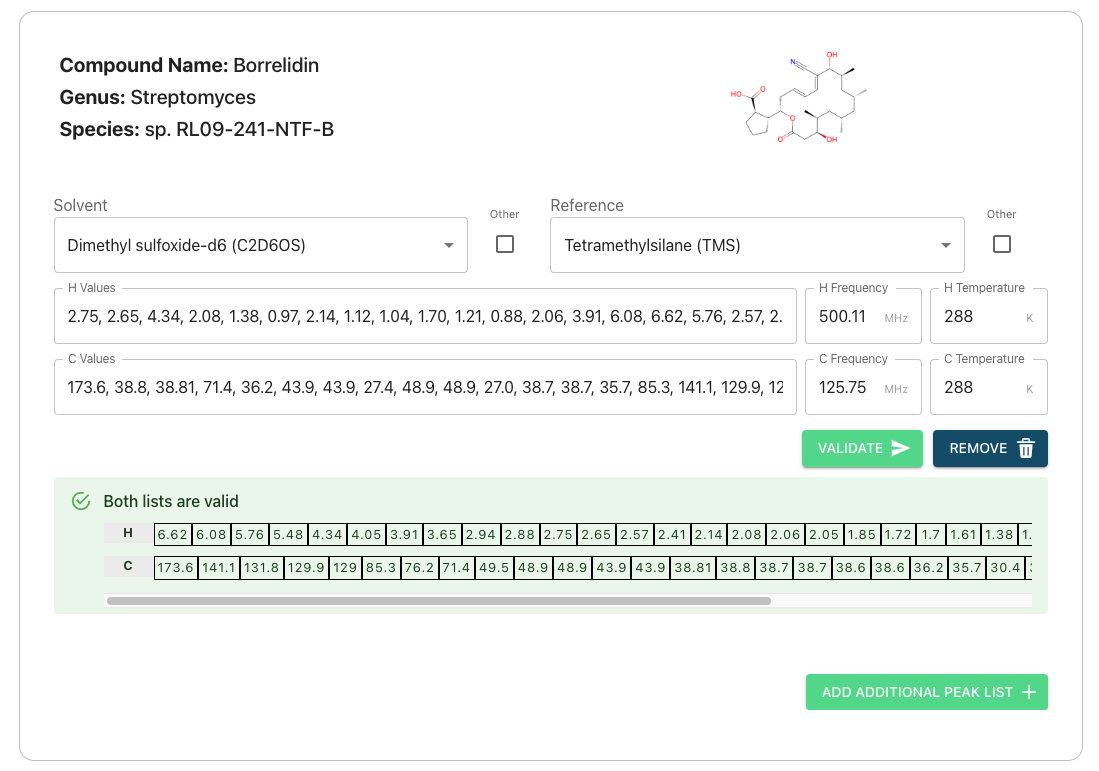
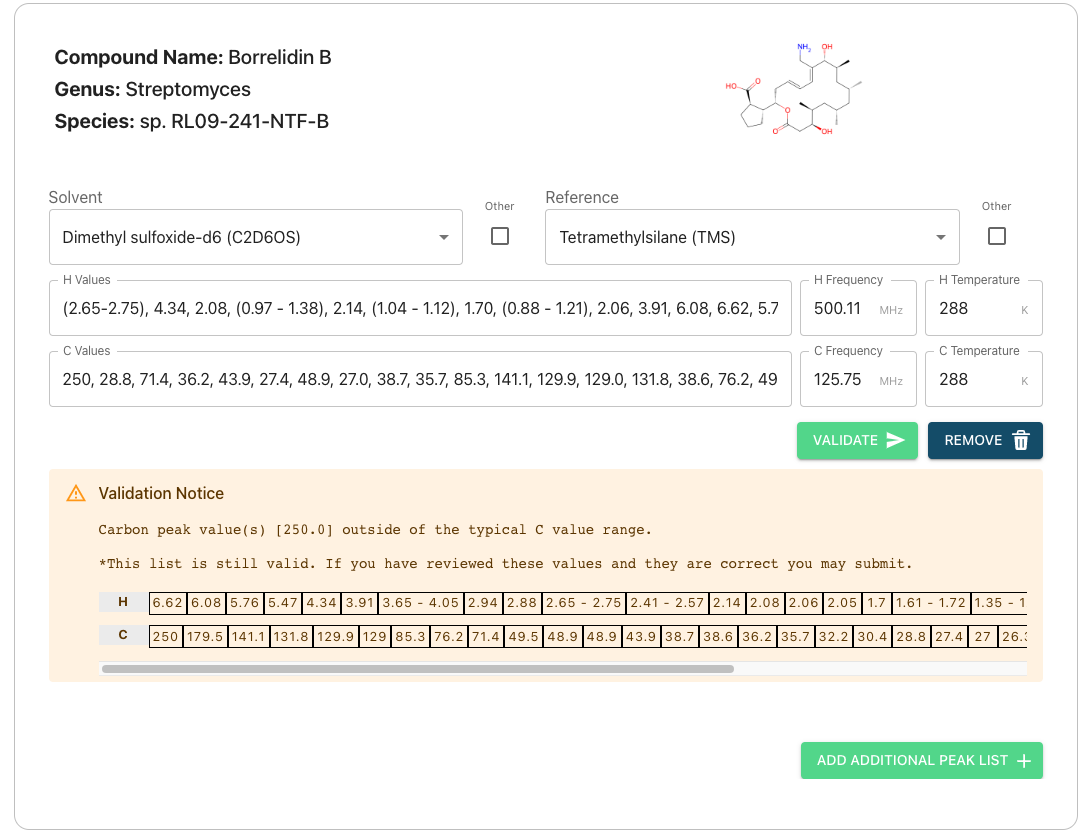
On the peak list submission page you will see the compound info you entered previously and some text boxes. The text boxes can be used to enter your peak lists for Carbon and Hydrogen atoms on a compound-by-compound basis.
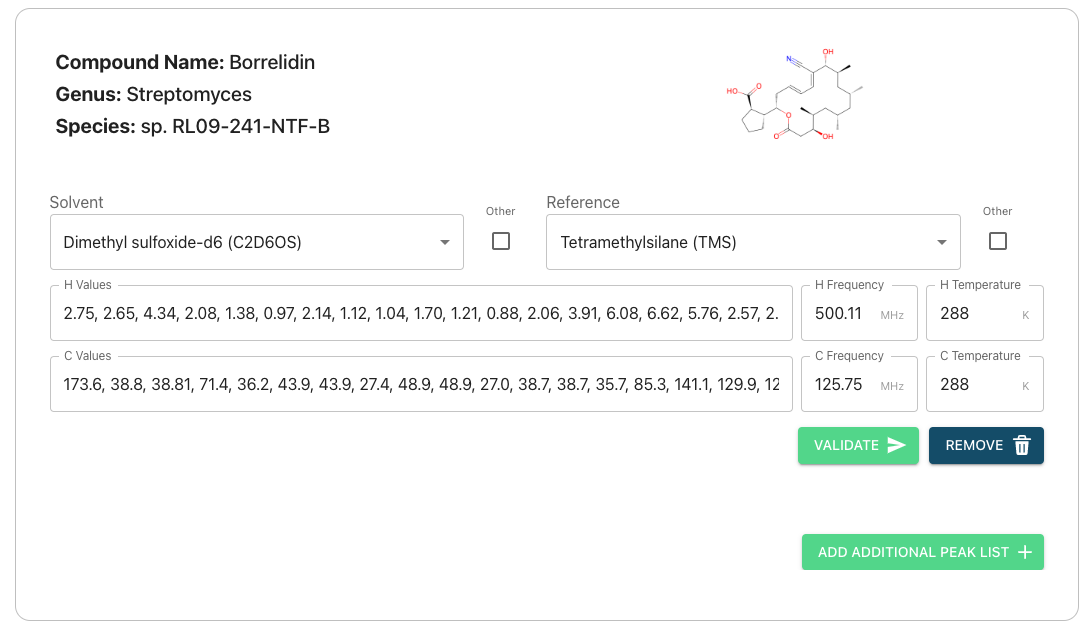
Autofilling Solvent, Frequency, and Temperature Information
-
If you have submitted nmr data then our system will try to extract solvent, frequency, and temperature information from your 1D experiment files. However, this takes time and it is currently possible to reach the peak list page prior to your data being fully processed by our system, in which this feature cannot be used. If you wish to utilize of it and your data does not autofill please wait and refresh your page. This may take several minutes if you have uploaded multiple compounds.
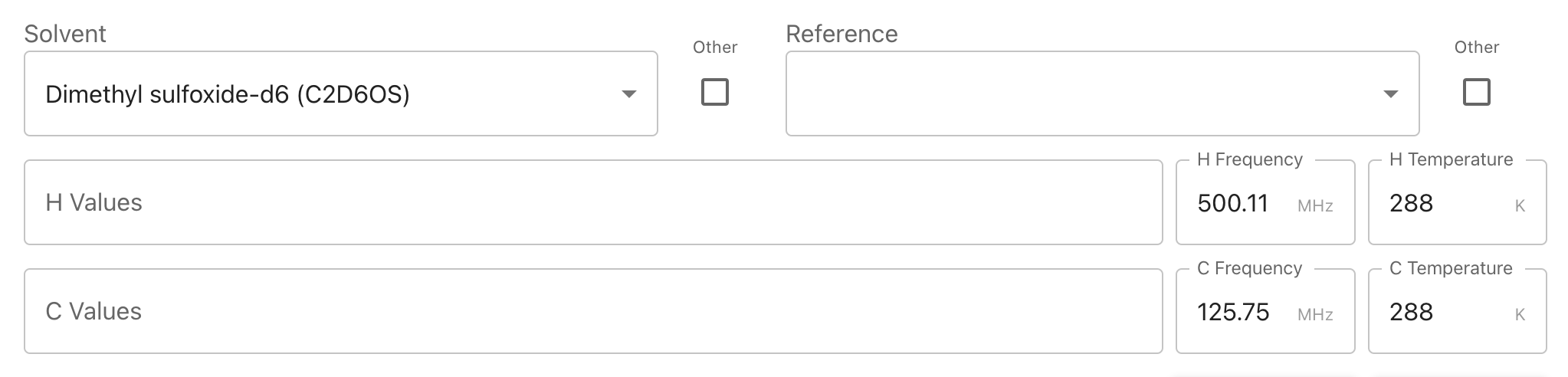
Submitting Other Solvents or References
-
If the solvent or reference you used is not listed in the dropdown you may check the “Other” box next to either. This will allow you to type your solvent directly. Please enter the chemical formula of your solvent rather than its name.

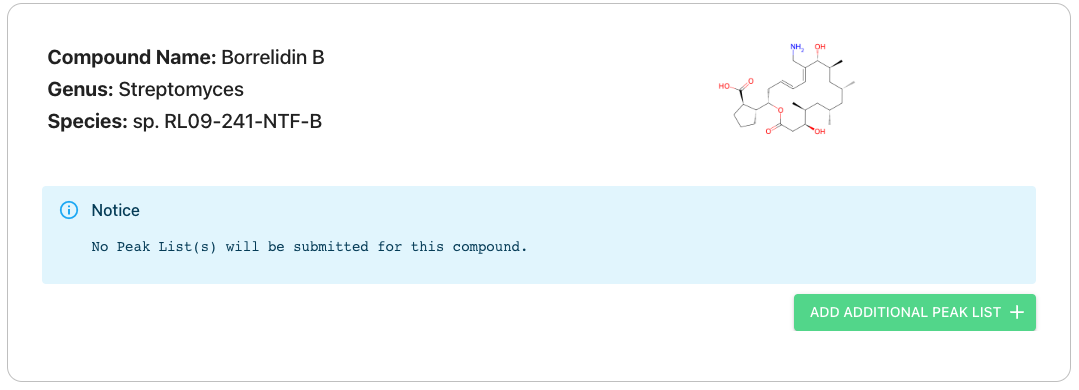
Skipping peak list submission for specific compounds
-
You can opt out of submitting a peak list for a specific compound by clicking the “Remove” button until there are no peak lists included for that specific compound. Note that if you have previously peak lists they will be deleted if you re-submit with no peak data.
Validating
- Clicking the validate button will cause our system to check if your submitted list is valid and provide you with immediate feedback underneath each compound
- You must validate with no Errors (red) before submitting
- Lists that throw warnings (yellow) or provide feedback information (blue) are still valid to submit. Please read the details and double check your peak lists to ensure they are correct.
Submitting
- Clicking submit will cause the system to validate. If it is valid you may proceed to the next step and your peak lists will be saved in our servers.