SNAP-MS Documentation
SNAP-MS is a tool for predicting natural product compound classes from mass spectrometry data. It works by comparing groups of masses derived from MS/MS fragmentation spectrum matching (e.g. molecular networking) to groupings of candidate molecules from the Natural Products Atlas based on compound similarities. Candidate compound classes are prioritized based on their coverage of the original MS/MS groups (i.e. how many of the mass features are accounted for by the candidate compound family).
SNAP-MS is described in the following open-access publication:
SNAP-MS is accessed through a dashboard on the NP Atlas website at:
www.npatlas.org/discover/snapms
Input Files and Uploading Data
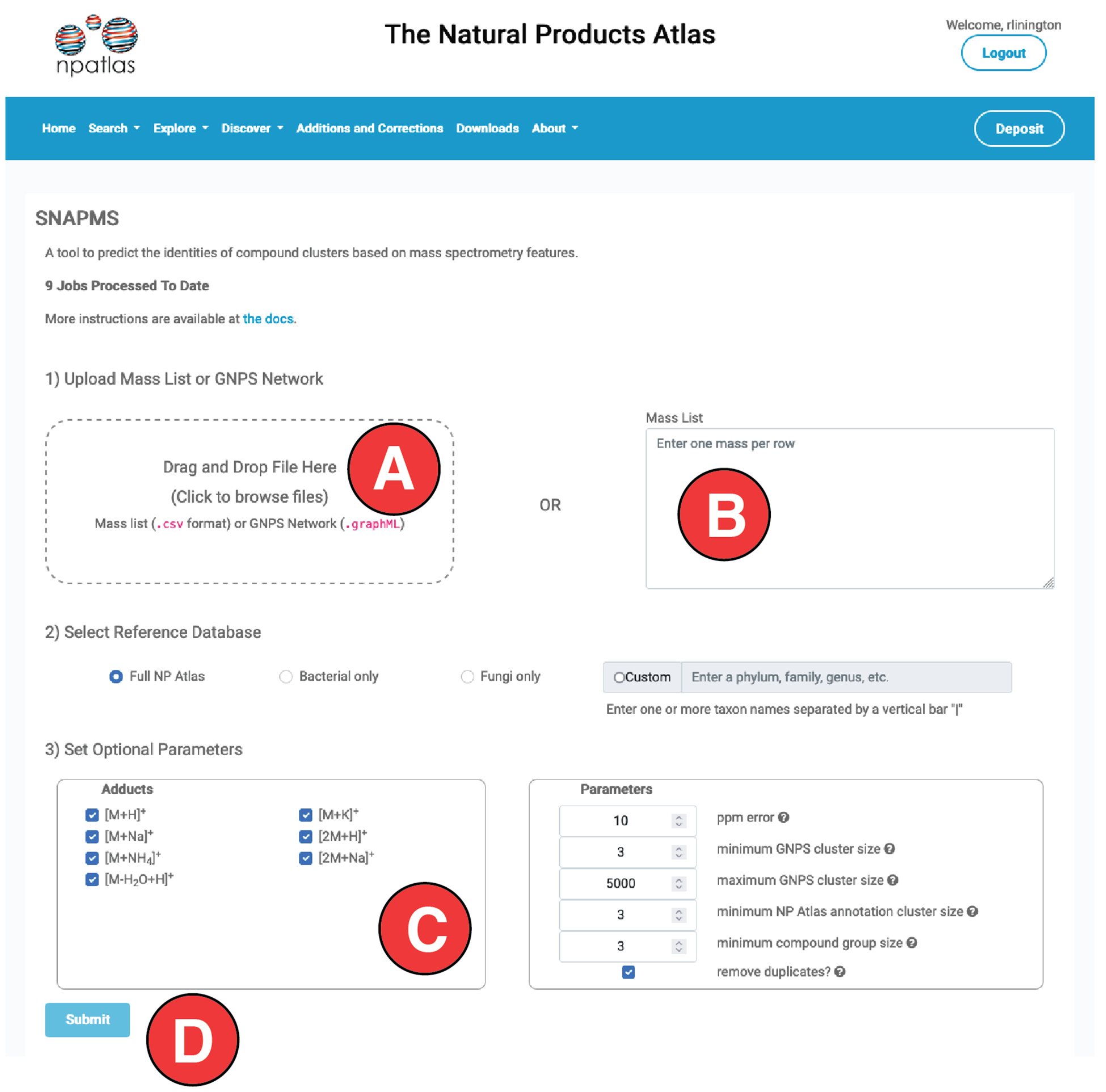
To use SNAP-MS simply drag and drop your MS data file into the drag and drop region on the webpage (A) or enter masses into the mass list text box (B), set any optional parameters (C) and hit submit (D). Two file types are accepted- graphML and CSV.
graphML
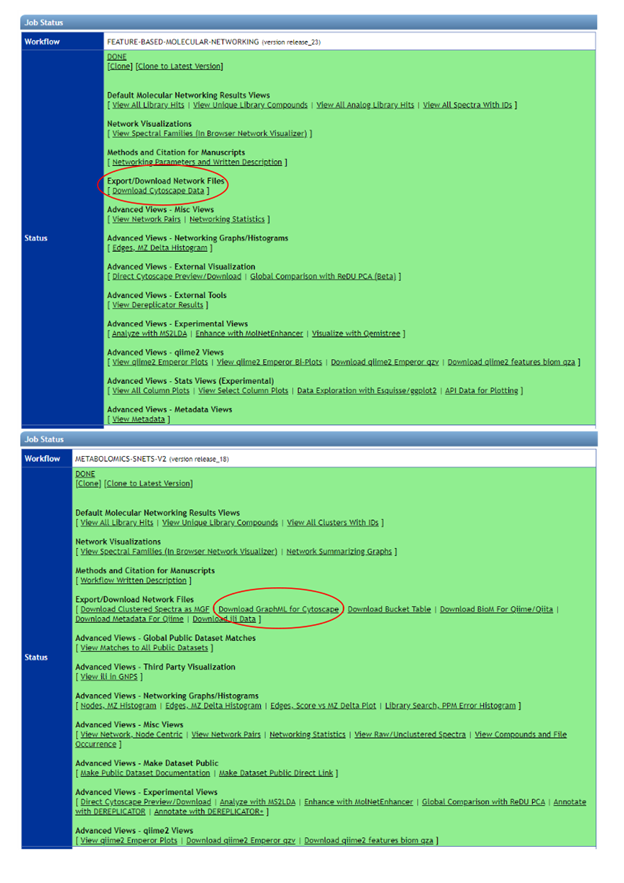
Output files from molecular networking may be analyzed directly in SNAP-MS by importing the molecular networking graphML file. Currently, SNAP-MS accepts files from both classical molecular networking and feature-based molecular networking.
NOTE: We currently (04/2024) do not support output files from GNPS2.
To export the appropriate file from GNPS navigate to your results page and select “Download Cytoscape Data” for feature based molecular networking or “download GraphML for Cytoscape” for classical molecular networking.
CSV Mass List
Any list of masses may be analyzed by importing a simple mass list as a CSV file. This allows users to analyze sets of masses from related HPLC peaks or other grouping methods, without the need to run molecular networking or other MS/MS fragmentation matching tools.
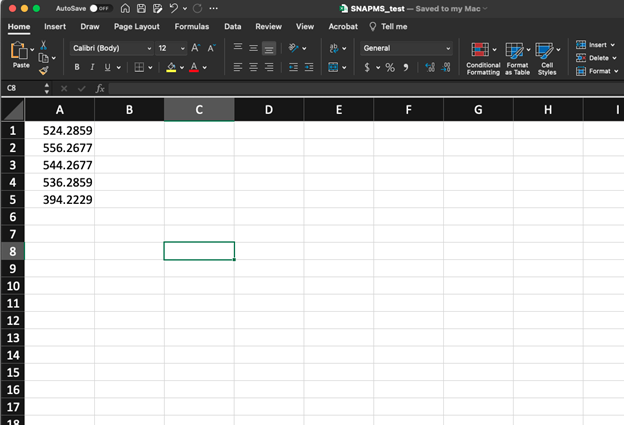
Users should create separate mass lists for each group of masses (i.e. you should run separate jobs for each compound class by submitting separate CSV files). The CSV file should contain a single mass on each line, without column headers or additional data in other columns. These can easily be created in Excel by placing a different mass in each cell of column A and selecting:
File-> Save as -> CSV UTF-8
Alternatively, users can enter masses directly into the text box in the top right (B) without the need to create a separate mass list file. Note that the original masses are not stored in the results file if the text box is used so it can be helpful to create mass list CSVs in order to stay organized about which mass lists were used to create which results files.
Analysis Parameters
SNAP-MS is configured with a set of standard default parameters that should be suitable for most situations. However, parameters can optionally be configured using the parameters panel.
Reference Database
By default, SNAP-MS uses the full Natural Products Atlas database as the reference. However, narrowing the reference database to include only compounds that are relevant to the search can significantly improve the accuracy of the tool. Options exist to select only the bacterial or fungal components of the Atlas database. We strongly encourage users to take advantage of this feature in their analyses. Alternatively, users may specify one or more taxa in the custom search box. These must be legitimate taxonomic terms (if in doubt, try searching for them in the Taxon box on the Basic Search page of the Atlas site: www.npatlas.org/search/basic). To specify more than one term, please use the vertical bar character | as the separator.
Note that the Natural Products Atlas currently reports the first instance of isolation rather than all instances of discovery, so filtering down to the genus level can sometimes omit compounds that were first isolated from other genera.
Adducts
A range of possible adducts are provided in the adduct section. If you know the adduct of your mass features you can reduce the number of candidate classes by limiting the adduct selection (deselect check boxes).
Ppm Error
By default the parts per million (ppm) mass error is set to 10. For instruments with lower or higher resolving power this can be modified. Note that SNAP-MS is designed to be used with ‘high-resolution’ mass data, and will not work well with data from ‘low resolution’ instruments such as single quads or ion traps.
Minimum GNPS Cluster Size
The minimum number of mass features present in a subnetwork for that network to be analyzed. We do not recommend reducing this value below 3 because the number of candidate families can become very large if the number of target masses is low.
Maximum GNPS Cluster Size
The maximum number of mass features present in a subnetwork for that network to be analyzed. Set to a very high number by default, this can be reduced if your network contains large subnetworks, which are typically composed of multiple compound families and do not analyze well using this method.
Minimum NP Atlas Annotation Cluster Size
Minimum number of compounds in a candidate SNAP-MS annotation for that annotation to be included in the results set. Recommended minimum number is 3, because including very low annotation cluster sizes can return large numbers of low confidence results.
Minimum Compound Group Size
Minimum nubmer of discrete mass features in a given subnetwork annotated by SNAP-MS for that annotation to be returned. In other words, if this is set to 3 then at least 3 different masses in the subnetwork must have annotations from the same compound family for that compound family to be returned as a candidate answer.
Maximum Results Nodes
Maximum number of nodes permitted in a single results graph. Used to exclude very large and uninformative graphs and prevent large increases in the size of the results file.
Maximum Results Edges
Maximum number of edges permitted in a single results graph. Used to exclude very large and uninformative graphs and prevent large increases in the size of the results file.
Remove Duplicates
Occasionally subnetworks from molecular networking can contain multiple nodes with the same or very similar masses. Including the same masses multiple times in the SNAP-MS analysis can distort the ranking of candidate answers solutions because this ranking is based on the number of nodes in the original subnetwork that are annotated by each solution. Checking ‘remove duplicates’ limits the mass list to only unique masses in the list, within the set ppm tolerance.
Submit Job
Once you have provided your mass data and selected the parameters, hit Submit. You will be directed to the results page for your job. NOTE: you need to bookmark this page if you wish to return to your results at a later date. The server status indicates the current state of your job. Small analyses (e.g. individual mass lists) usually complete in a few seconds. Large molecular networks may require more time to complete, making it important to save the bookmark so that you can access the results once the job is complete.
Results
Each results page is found at a unique url that is provided to the user during the submission process. Although these urls are public they are complex codes which are not easy to guess, meaning that it is not possible to browse results files without knowing the results url. The results page provides a summary of the submission parameters, including the name of the file used for supplying the mass list(s) and the date of submission. Results are retained for 30 days and then deleted to save server space.
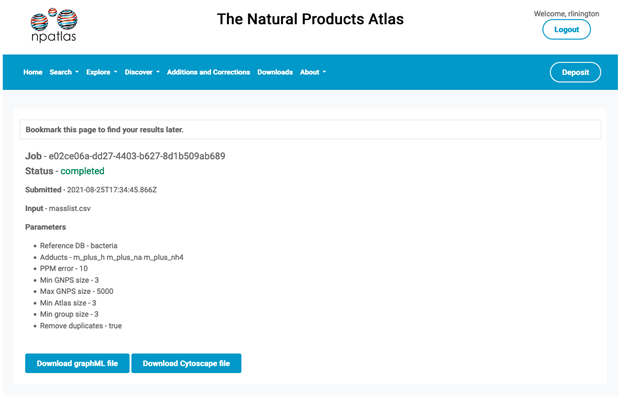
Download
The Download section on the results page offers two options: graphML or Cytoscape session file (.cys). The structure of these two files differs depending on which data format was originally submitted. If the original data was a CSV mass list or masses in the mass list box then the graphML and Cytoscape files both contain a single graph containing the candidate molecular families as separate subnetworks. Alternatively, if the original data was a molecular networking graphML file then the graphML download is a zipped folder containing a separate graphML file for each molecular networking subnetwork for which candidate compound families exist. Alternatively, the cytoscape results file includes one collection containing all of the results graphs. In both cases, results are related to the original molecular network via the ‘componentindex’ number, which is different for each subnetwork in the original molecular network file.
Interpretation
SNAP-MS predicts compound family identities from groupings of mass features and rank orders the predicted compound families based on the degree of coverage of the original mass lists. Importantly, SNAP-MS does not use MS2 data to determine the identities of individual molecules, and cannot differentiate between isobaric species for annotated nodes. Therefore, SNAP-MS results should be used to characterize the chemical composition of samples sets at the class level, but not to identify the structures of individual nodes. Compound identification requires additional inspection and assessment of the original MS data, as well as other spectroscopic data that may be available (NMR, UV, etc).
Secondly, all candidate classes that meet the selection criteria (minimum NP Atlas cluster annotation size etc.) should be considered as potential solutions and evaluated independently. In the Cytoscape results files the top ranked candidate(s) are indicated by a thick blue node edge for all subnetwork members.
Limiting the reference database to only include relevant taxa can greatly improve the accuracy of the predictions by eliminating candidate solutions from unrelated source types (e.g. fungal compound classes as candidate solutions for masses from a Pseudomonas extract library). In addition, modifying the ppm error and other parameters to values appropriate for your instrument can improve both coverage and accuracy of the candidate compound families.
Support
For additional help with questions related to SNAP-MS please email snapms@npatlas.org
![]()